About the science
Alzheimer's disease (AD) and other neurodegenerative diseases as lysosomal protein storage diseases
The true underlying cause of AD has eluded science to date, but substantial learnings about pathophysiologic processes and the role of amyloid accumulation in some cases has increased our understanding. Treating the underlying cause of AD and other diseases will be critical to making ultimate progress in this health care crisis.
AD and other neurodegenerative diseases that accumulate amyloid-like proteins have many clear parallels with lysosomal storage diseases that affect the brain. These scientific parallels include their biochemistry, histology and pathophysiology including amyloid accumulation, macrophage capping neurons, tauopathy features and neurologic features like damage to the hippocampus with memory impacts (Table 1). In totality there appears to be a strong correlation between lysosomal dysfunction and amyloid accumulation. Lysosomal biogenesis is maximally activated in AD brains at death consistent with the lysosomes being in a dysfunctional state and consistent with the pattern expected in a lysosomal storage disease (Nixon et al).
Treating AD models with increased PPCA via gene therapy can reduce or prevent amyloid plaque. We have studied the idea that the accumulation of amyloids both extracellularly and intracellularly is due to a relative deficiency of proteases needed to cleave the very difficult to digest oily protein oligomers and fibrils that accumulate with age, particularly within the lysosomal compartment. Work by Dr. Alessandra d’Azzo at St. Jude Children’s Research Hospital in Memphis and our own research team have now demonstrated that a protease PPCA can efficiently cleave Ab amyloid in monomeric or oligomeric forms in vitro. By delivering supplemental amounts of this lysosomal protease to the brain via AAV gene therapy in murine models of AD, we have also shown that the increased PPCA enzyme levels in the lysosomes of neurons reduces amyloid plaque accumulation outside the cell more effectively than monoclonal antibodies targeting amyloid (Aβ42). This is precisely because clearing the amyloid and toxic oligomers contained within the neuronal cell lysosomal compartment reduces the rupture of neurons and exocytosis of undigested amyloid outside the cell where it can become plaque.
This insight is a fundamental change in understanding of how and why amyloids accumulate and cause disease and opens the door to a profound single-shot treatment to reduce or prevent AD.
| Lysosomal Disease (Gene) | Type of Amyloid Deposition | Observation |
|---|---|---|
| Batten Disease/CLN2 (TPP1) | Amyloid like proteo-lipid material | Extracellular Aβ-amyloid accumulation and neuronal cell death in patient brain |
| Gaucher disease (GBA1) | α-synuclein | α-synuclein accumulates in Lewy bodies and neurites in brain & spinal cord of patients leading to Parkinsonian symptoms |
| MPS IIIA & MPS IIIB (SGSH & NAGLU) |
α-synuclein Phospho-Tau Increased Aβ-amyloid |
Amyloid protein aggregates detected in patient postmortem brains. In addition, aggregation of amyloid proteins in neuronal cell bodies in mouse model brain |
| Sialidosis (Neu1) | Aβ-amyloid | Extracellular amyloid accumulation in Neu1-/- mouse brain |
Delivery of PPCA enhances lysosomal function and degradation of amyloid
Protective protein cathepsin A (PPCA) has two distinct functions, both of which serve to break down amyloid aggregates:
- In the endosomal/lysosomal compartment, PPCA has carboxypeptidase activity at acidic pH directed towards substrates with hydrophobic amino acids.
- PPCA stabilizes neuraminidase 1 and beta-galactosidase to form a multi-enzyme complex, actively enhancing lysosomal clearance of proteins.
AAV9 PPCA gene therapy in murine model of severe AD
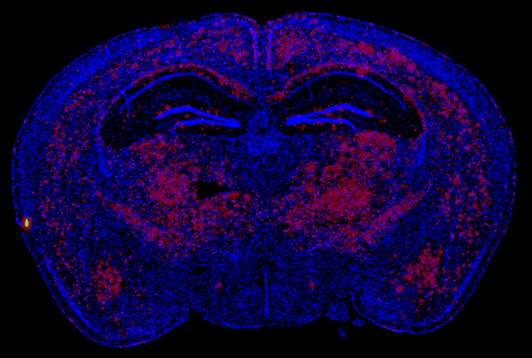
Vehicle treated
PPCA treated
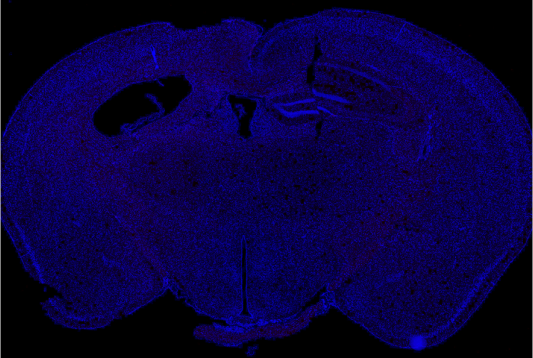
12 weeks following AAV delivery of PPCA we see near complete reversal in an aggressive murine model of severe AD (5xFAD), with the extent of clearance correlating with PPCA brain exposure. (Red=Amyloid)
Vehicle treated
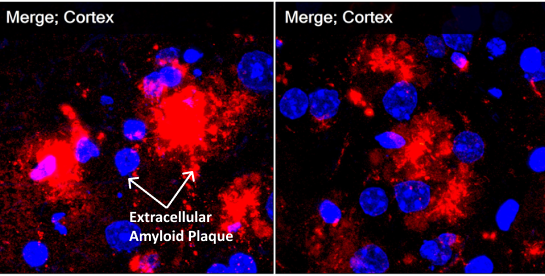
PPCA treated
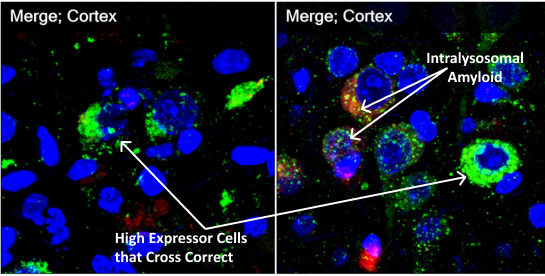
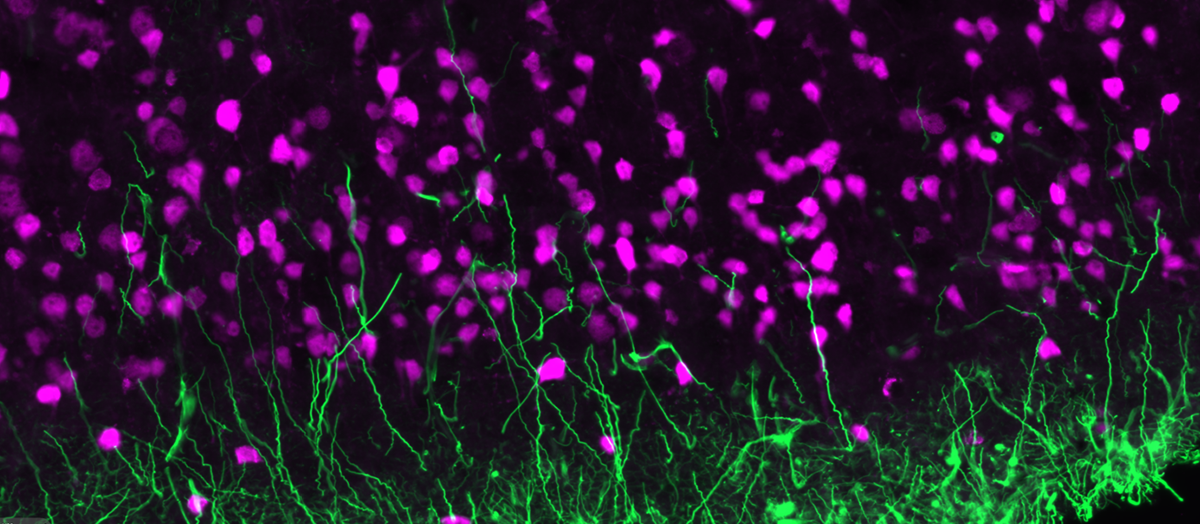
Immunohistochemistry staining shows massive fluffy red-staining amyloid in control vehicle-treated murine models outside cells but also in the perinuclear lysosomes. On the right in the treated animals, green-staining PPCA is in the perinuclear lysosomes with tiny pieces of red amyloid (now showing yellow in the merged pictures) colocalizing with PPCA. These pictures show that lysosomal PPCA can be cross-corrected from high expression cells, increase PPCA in many neurons and enhance lysosomal degradation of amyloid. (Red=Amyloid; Green=PPCA)